``
Hello and welcome to week 6 of ESPM 112L-
Metagenomic Data Analysis Lab!
- What’s required to perform phylogenetic analysis?
- What software will we be using to get this info?
- Getting your sequences
- Aligning your sequences
- Visualizing your alignment (local system)
- Creating a phylogenetic tree (back on the cluster)
- Visualizing your tree
This week we’re going to be going over some material that’s very near and dear to my heart… phylogenetics!
A lot of microbial ecology research revolves around phylogenetic analysis, especially my own. So what is it, exactly?
Phylogenetics is basically the process by which we estimate relationships between organisms. In the case of today’s lab, we’ll be using it to measure the relationships between bacteria in your baby gut samples.
What’s required to perform phylogenetic analysis?
- A sequence set (DNA or protein)
- A multiple sequence alignment program
- An alignment (made by using the two previously mentioned items)
- Phylogenetic tree estimation software (FastTree, iQ-TREE, RAxML, etc)
- Phylogenetic tree visualization software (iTOL, FigTree, etc)
What software will we be using to get this info?
- Sequence set: ggKbase
- Multiple sequence alignment: Mafft and FAMSA (you'll only use MAFFT today)
- Mafft: https://mafft.cbrc.jp/alignment/software/
- FAMSA: https://github.com/refresh-bio/FAMSA (Wicked fast!)
- Alignment viewer: Aliview (https://ormbunkar.se/aliview/)
- Tree building: FastTree (on cluster)
- Tree visualization: iTOL (http://itol.embl.de)
Getting your sequences
Log in to class.ggkbase.berkeley.edu on your browser, navigate to your cow’s project page, and click “Genome Completeness -> Ribosomal Proteins” near the top of the page. You’ll see a menu that looks like this:
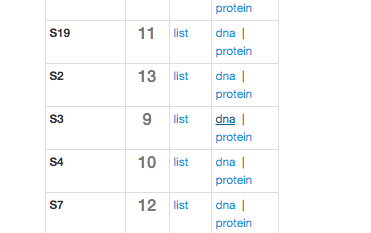
Be sure to select rps3- it’s generally a very solid marker gene, used for similar purposes to 16S rRNA. Click “Protein” and a file will be generated and downloaded to your computer. Now you’re going to want to put that file on the cluster with cyberduck or scp.
Aligning your sequences
Now log in to class.ggkbase.berkeley.edu on the terminal. Again, make sure you’ve put your sequence file you’ve downloaded from ggKbase onto the class server! (If you would prefer not to load things on the cluster, talk to me. There are online versions of these tools.)
Mafft is a popular aligner renowned for its accuracy and thoroughness when creating alignments. It takes a while, though, since it was written with accuracy in mind. Try aligning your sequences with mafft:
mafft --localpair --maxiterate 1000 --reorder --thread 4 rps3.faa > rps3.mafft.mfaa
This alignment should take around 4-5 minutes; if it runs longer, don’t be too concerned.
Remember to choose a name for your alignment and to end that filename with .fna or .mfna. I prefer to use .mfna because that means (to me) that it’s a multiple sequence alignment (m for multiple) of nucleotide sequences (fna for fasta amino acid). Just makes things easier later. A protein multiple sequence alignment, for example, would be .mfaa. It makes it easier to identify what’s in your files, since a FASTA file can be DNA, RNA or protein!
Visualizing your alignment (local system)
On your local computer, install Aliview, which we’ll be using to view your alignment: https://ormbunkar.se/aliview/
Now use cyberduck or SCP to download your multiple sequence alignment to your computer. Open Aliview, and use it to open this multiple sequence alignment file. What do you see? Are there regions of conservation? Is it particularly gappy? This is just to give you an idea of what your data looks like and what you’re actually working with.
You may want to delete partial sequences- i.e. rows that are comprised primarily of gaps- before building your tree.
It’s common practice to trim your alignment before building a phylogenetic tree- that is, to remove columns from your alignment that consist primarily of gaps. This often helps to construct more reliable and robust trees, but the best way to do this is subject of debate within the field. It’s also quite common to trim specifically at the beginning and end of the alignment.
Creating a phylogenetic tree (back on the cluster)
Now that you’re back on the cluster, make a folder to do your tree analysis in. Phylogenetic tree building software gets a little messy sometimes and we don’t want to get confused about what files go where. It’s good to keep your home directory tidy.
Make a directory for this analysis (with mkdir) and move (mv) your DNA alignment into this folder. Now navigate in there (cd).
You’re going to be using FastTree as your tree building software today- here’s an example command.
fasttree rps3.mafft.mfaa > rps3.mafft.mfaa.treefile
This should be very fast- less than a minute.
Now remember, when you use >, you’re creating a new file! So the above command is creating a file called rps3.mafft.mfaa.treefile. (You can name that file whatever you want, by the way, I just want to be consistent for the purposes of the tutorial.)
Visualizing your tree
One of the best ways to visualize trees is with a website called iTOL. This site has recently moved to a subscription service, so you don’t get all the functionality with a free account, but it’s perfectly sufficient for our purposes today. You’re going to make an account, upload your tree, and send it to me as part of your turnin today.
Uploading your tree to iTOL
First, download your treefile to your local computer using cyberduck/SCP. Then, go to and click ‘Register’ at the top right of the page to create an account.
Once you’ve created an account, log in; this will take you to a page containing two buttons, “Paste tree text” and “Upload tree files”. Click “Upload tree files” and upload the tree file you just downloaded from the class server.
Interpreting your trees
For those of you not in the lecture, we’ve talked a lot about trees, and so if you don’t have experience interpreting these, you might be at somewhat of a disadvantage- call me over at this step if you’d like to learn more about what’s going on here.
I will do a live demo of this in lab, and come around for help, since this might be the tricky portion of today’s lab.
First, now that you’ve uploaded your tree file, click on the link and go view it. You should see a circular representation, with a bunch of impossible-to-read small text- don’t worry about that just yet.
There’s a box at the upper right of the page that looks like this:
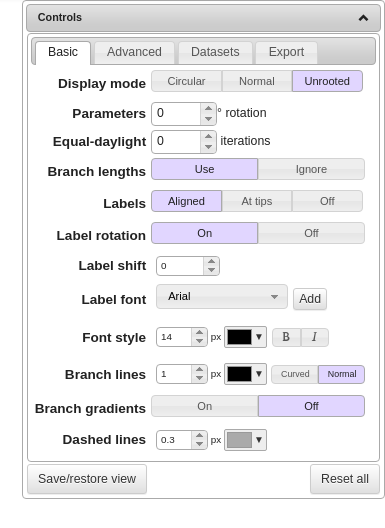
Select “Unrooted” (as in the example above) and the tree should look like this:

Finding cool stuff in your tree
Find a branch that particularly interests you. The vast majority of your sequences will be from bacteria, which shouldn’t surprise you- but there’s more than just bacteria in here.
Click on the branch you’d like to investigate, or the text label for that branch (call me over if you have trouble with this), and click the blue text at the bottom of the resulting window, like so:

As an example, the branch I copied looks like this:
scaffold_383062_77|metabat_scaffolds2bin_tsv_074|JS_HB1_S134
But we just want the part before the first | character, i.e.
scaffold_383062_77
Go back to class.ggkbase.berkeley.edu, and in the top left search bar, paste that sequence ID…. what’s the taxonomy for that protein? Go to the contig- is it in a bin?
See if you can find an archaeon among one of your longest branches!
Today’s turnin:
-
Send me the URL for your tree.
-
Search at least three proteins on ggkbase using the method I described above using scaffold_383062_77 as an example and send me the taxonomy for each one.
-
How many columns are in your alignment? (Open it in aliview on your local machine and scroll all the way to the right!)