``
Hello and welcome to week 12 of ESPM 112L-
Metagenomic Data Analysis Lab!
This week we’re going to do two things:
-
Talk about your projects (In class, not on the walkthrough)
-
Learn how to run iRep, including making an alignment
Let’s get started with iRep!
iRep
iRep is software developed to estimate the growth rate of bacteria by using genomes obtained from metagenomes. iRep looks at the reads which align to that genome, and based on the pattern of coverage it displays, estimates how quickly that population of bacteria is replicating their genome.
The reason this is possible is due to the way in which bacterial cells replicate their DNA. Remember, most bacterial chromosomes are circular. They replicate their genome starting at a central point - the ‘origin of replication’- and finish at the ‘terminus’. If a population is growing quickly, more reads will be generated from the area near the origin, since that’s where everyone is making new DNA.
iRep produces an ‘iRep value’ as its most important statistic. This ranges between 1 and 2; an iRep value of 1 means that the population currently isn’t experiencing any replication. An iRep value of 1.5 means that half the population is estimated to be currently dividing. And iRep value of 2 means that the entire population is currently dividing.
In order to use iRep, we need to make an alignment between the reads and a reference genome of your choice.
First step: Choosing a good genome from your sample
Go to class.ggkbase.berkeley.edu and log in; find a nicely sized genome (i.e. >= 1.5Mbp) with a reasonably low number of contigs, like the example below.
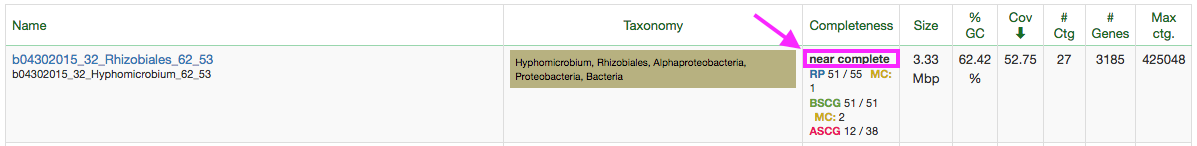
Now you can find that bin at /class_data/assemblies/cow_bins/[YOUR SAMPLE ID]/[BIN NAME].fasta. Copy that to your home directory.
Second step: Making a directory to perform your alignment
It’s preferable that one person per group do this, but it’s ok if more than one person does. You’re going to generate a lot of .sam files doing this, which contain your alignments. These files are huge, so once you’re done with today’s lab be sure to delete them!
Let’s make a folder in your home directory and call it iRep.
mkdir ~/iRep
cd ~/iRep
Now you want to move the fasta file you chose into this folder. Use either cp (to copy) or mv (to move) that file into this directory. (i.e. cp ../example_genome_bin.fasta .)
Now that you’ve got that set up, let’s get the read files from your sample and put them here. These files, again, are huge, so we don’t want to copy them over- we’re going to be using the symbolic link trick again today. (If you don’t remember what this is ask me and I’ll be happy to explain.)
ln -s /class_data/reads/[your sample ID]/raw.d/*trim_clean.PE.*.fastq.gz .
This creates links to the read files, so that you can access them as if they were in your current directory, but you haven’t made a duplicate copy so we can save some hard drive space.
You’ve got all the pieces put together! Now let’s make an alignment.
Third step: Making an alignment
You’re doing great. You’re almost at the point where you can have a breather. We’re gonna make an alignment; the key difference between this week’s job and what you did last time is that this week we’re generating a .sam file rather than a .bam; we’ll discuss the differences in class, but be aware that sam files are MUCH larger than bam files and you should delete it when you’re done using it!!!
Here’s how to do the alignment:
#Remember, you're in the directory ~/iRep.
#Actually perform the alignment. This should take ~6-10 minutes.
bbmap.sh ref=[GENOME FASTA FILE] in1=[FORWARD READ FILE] in2=[REVERSE READ FILE] threads=4 pigz=t unpigz=t ambiguous=random out=stdout.sam | shrinksam > genome_alignment.sam
Which files are the forward and reverse read files, I hear you asking? Well, you should see two .fastq.gz files; one ends in .1.fastq.gz, and is your forward reads file; the other ends in .2.fastq.gz and is the reverse reads file.
You did it! That was the hard part. Let’s run iRep.
Fourth step: Running iRep
This part, while it might take a few minutes, should be relatively easy in comparison to what you’ve already done. First, try running iRep -h and read the options. Feel free to edit some of these, but make sure not to edit the number of threads you’re using (just use 6 at a time please).
Here’s an example command:
iRep -f [genome bin fasta] -s genome_alignment.sam -t 6 -o iRep_output
This will create two files: iRep_output.tsv and iRep_output.pdf. Go ahead and download iRep_output.pdf, and take a look. What is the iRep value for this genome? What proportion of the population is estimated to be dividing?
Today’s turn-in assignment
Use another set of reads in /class_data/reads (i.e. not your sample) and align them to your genome just like we did above with your sample’s reads. Generate another .sam file (make sure not to give it the same file name as your first one!) in the same way as before. Run iRep again, but change the command slightly;
iRep -f [genome bin fasta] -s *.sam -t 6 -o iRep_output_combined
The * is called a ‘glob’, or a wildcard character. Essentially, you’re telling iRep to take all files ending in .sam as input; *.sam basically means ‘anything.sam’. If that makes sense. (If you don’t understand this call me over and I’ll happily explain it to you in more detail.)
To turn in:
- Take the
.pdffile generated by this command and send it to me. - What are the iRep values for this genome in each of the samples you looked at?
- Is more of this population estimated to be replicating in your sample, or the second sample you looked at?
GOOD JOB! YOU DID IT!
